Explore the offer
Theory & Simulation / THEO & SIM
ATOMS AND MOLECULES IN MOTION
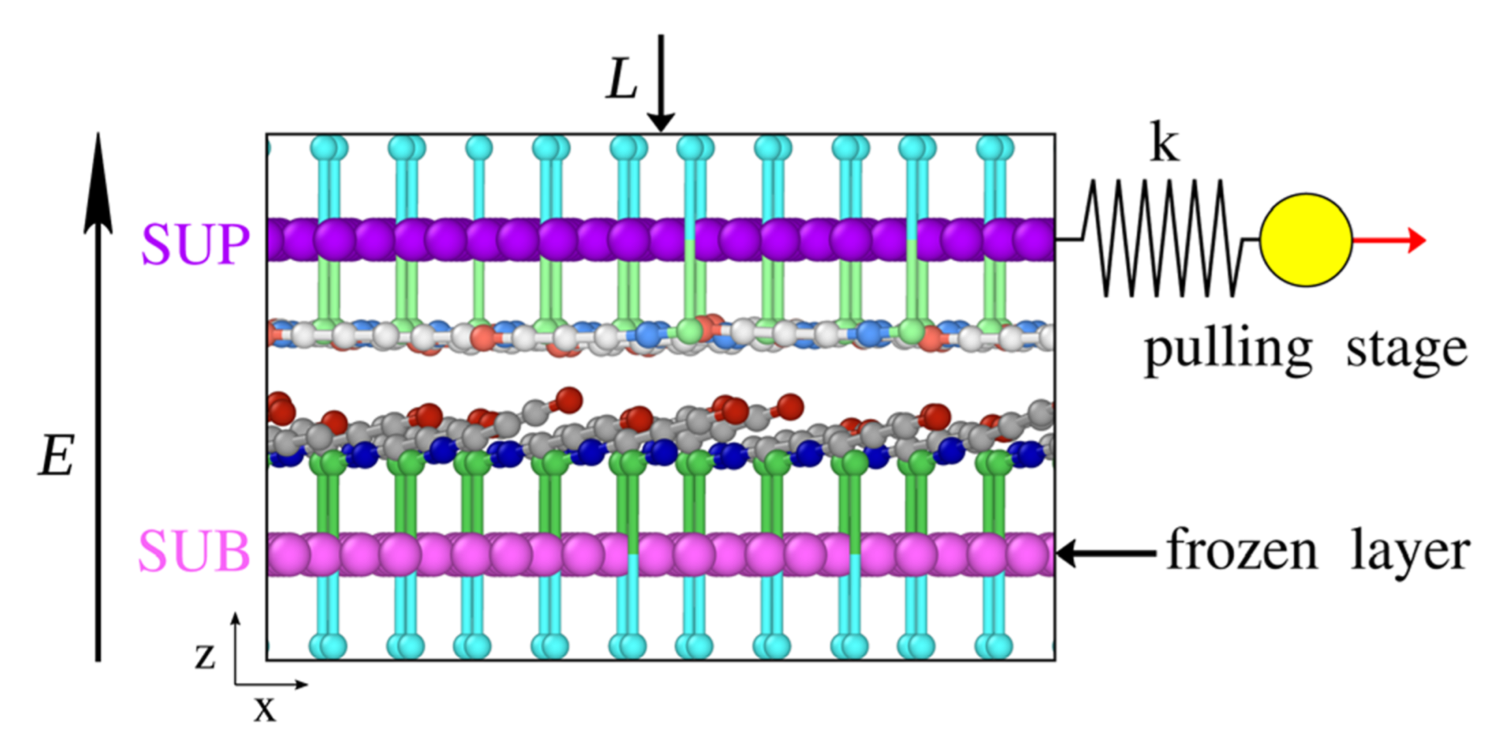
- Ab-initio molecular dynamics.Trajectories obtained from the Car-Parrinello Lagrangian or from the Hellmann–Feynman forces calculated on the Born–Oppenheimer (BO) surface within DFT calculations; several thermodynamic ensembles (NVE, NVT, NPT).
- Temperature activated events.Location of transition states, minimum-energy paths (Nudged Elastic Band balculations) and free energy landscapes (Metadynamics, constrained dynamics); Transition state theory and rates.
- Modelling the effects of complex chemical environments on a quantum-mechanical system.Quantum Mechanics/Molecular Mechanics approaches; Implicit solvents; Cavitation and pressure effects.
- Reactive Force Field Molecular Dynamics. Trajectories of thousands of atoms over time scales exceeding the nanosecond, simulated by force fields including chemical reactions (ReaxFF, eFF).
- Atomistic and Coarse-Grained Force Field Molecular Dynamics. Simulations of molecular and supramolecular systems formed by thousands-millions of atoms over time scales of microseconds using atomistic, residue-based or shape-based coarse-grain force field models.
Available instruments
Select instruments to view their specifications and compare them (3 max)
Lab's Facility
Milano